The Graphical User Interface
The graphical user interface (here) contains examples for RNA, DNA, and other nucleic acid anaolgs. The available examples generates similar structures to those published in the software article. They use a variety of nucleobases, backbones, helical parameters, and algorithm options. You can switch between these examples using the Input File option. You can run these examples as they are or you can use them as starting points for customizing your options. Bring the mouse pointer to the question marks ? in front of each option to get some explanation of the option.
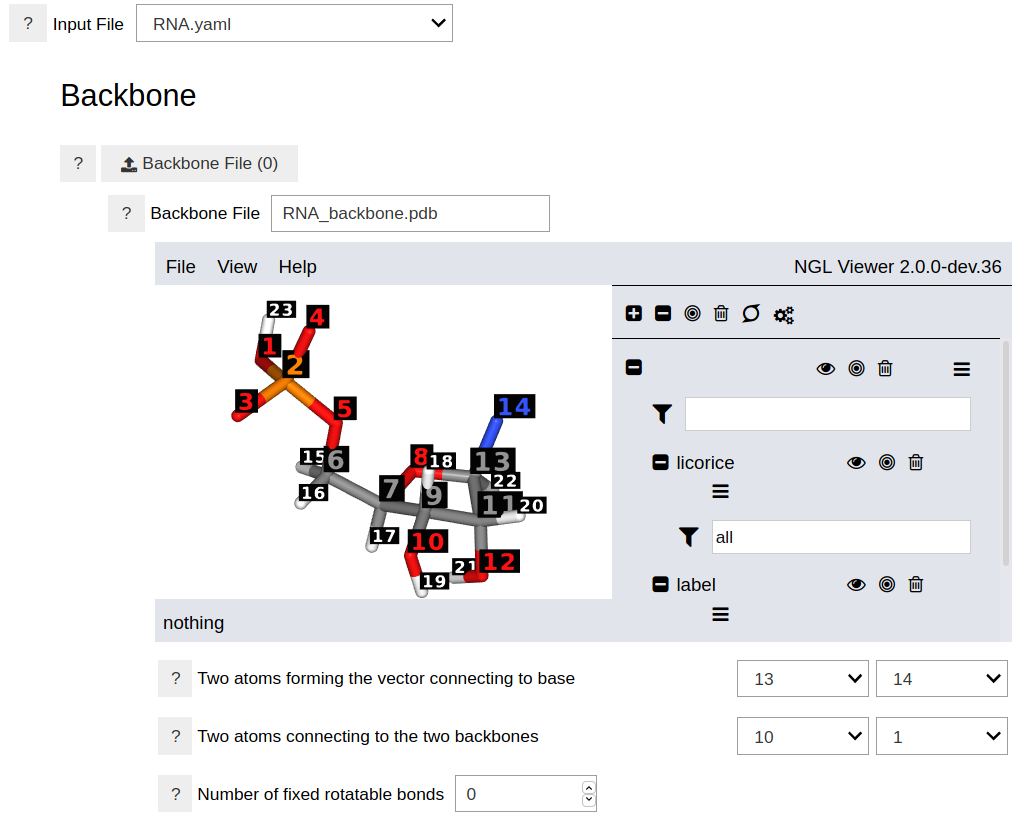
After choosing or uploading the input file, the graphical interface will display the backbone and nucleobase molecules and the other options.
The options are divided into four sections:
- Backbone
- Nucleobases
- Helical Parameters
- Runtime Parameters
As shown in the above figure, specifying the backbone requires three parameters: A path to a file containing the three-dimesional structure of the backbone, two indices specifying the connection to the nucleobase, and two indices specifying the connections to the adjacent backbones. The structure of the backbone can be uploaded, and many formats (e.g. PDB) are accepted. Optional parameters include the indices of fixed rotatable bonds. This is useful when the number of rotatable bonds in the backbone are large and the search space need to be limited to specific dihedral angles.
Uploading the geometries for the nucleobases may not be necessary. The program contains a library of the canonical nucelobases: adenine (A), guaninie (G), cytosine (C), uracil (U), and thymine (T). It also contains two alternative nucleobases that can adopt the hexad geometry, namely, aminopyrimidine (E) and cyanuric acid (I). If the user wants to construct a nucleic acid analog with alternative nucleobases, then the additional nucleobases must be defined as shown in the figure. The ZP.yaml example file shows how to define new nucleobases. The Z nucleobase is defined below.
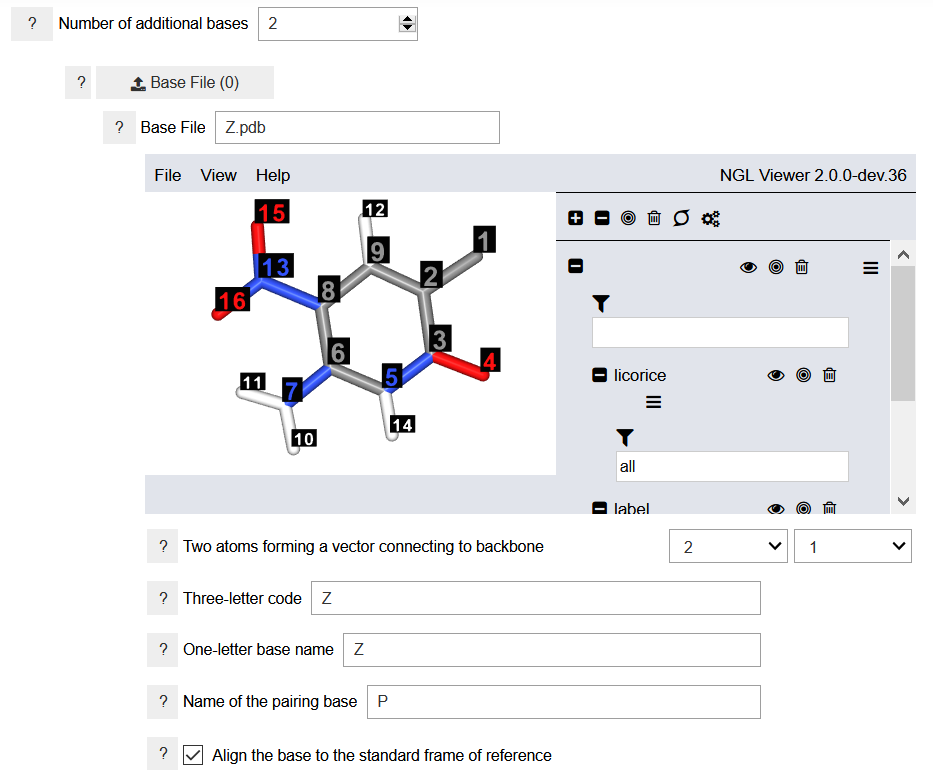
To specify the nucleobases, a three-dimensional geometry of the nucleobases must be provided. Additionally, two indices of atoms connecting the nucleobase to the backbone must be specified. The up-to-three-letter code is used to identify the residue name in the generated PDB files. The one-letter base name is a unique name that will be used in specifying the sequence of nucleobases in the strand. It should be different from the one-letter names of the nucleobases already defined in the program library. The name of the pairing base is the one-letter code for the pairing nucleobases. It is necessary when more than one strands are constructed. The coordinates of the specified nucleobases must be in the correct reference frame. This can be done automatically by the program, but it may need to be verified by the users. The new nucleobases are aligned to the DNA/RNA frame of reference. The program cannot align the nucleobases to a hexad frame of reference.
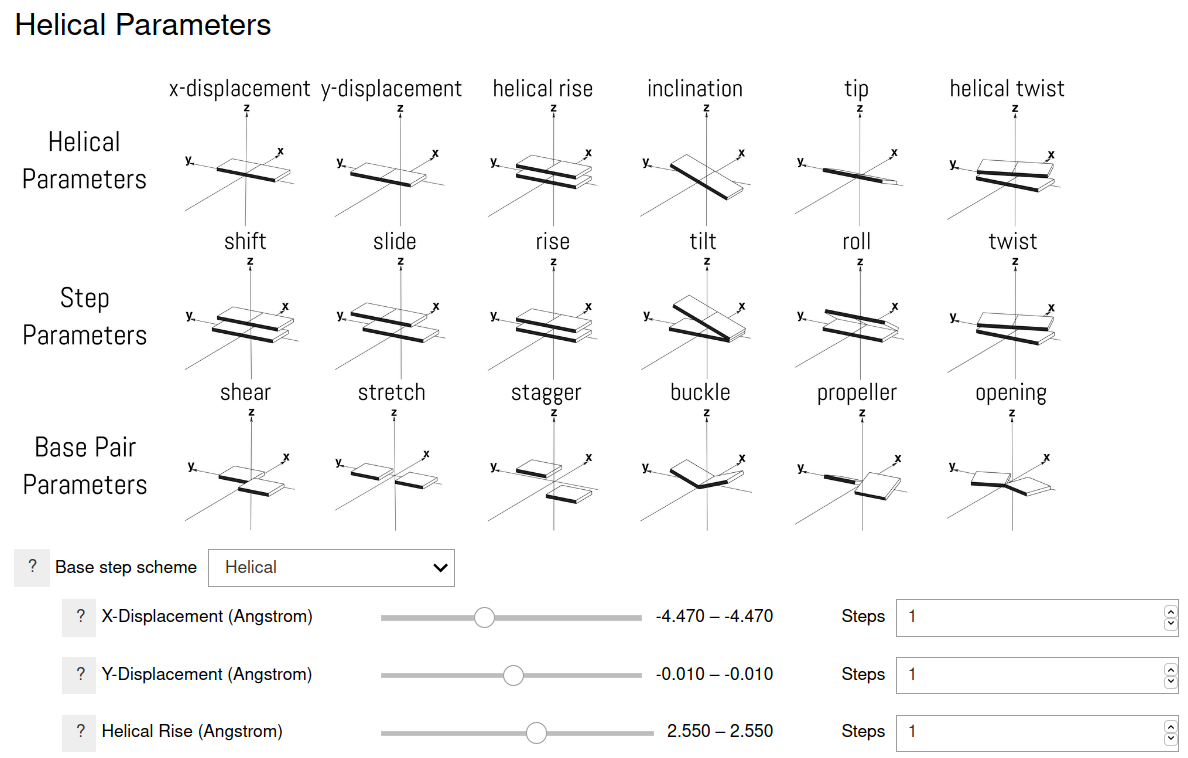
Six parameters specify the helical structure of the nucleic acid strands. Two equivalent schemes can be used according to whether a global helical frame or a local frame is used. Either scheme can be used for building the nucleic acid structure. Additional six parameters are used for speicifying the relative orientation of the bases in a base pair. All these options can be specified in the program. Single values or multiple values can be specified in a range. When a range of values is specified, the values are uniformally spaced between, and including, the the end points, with the specified number of configurations.
The runtime parameters include the options for specifying the conformational search algorithm and structural parameters.
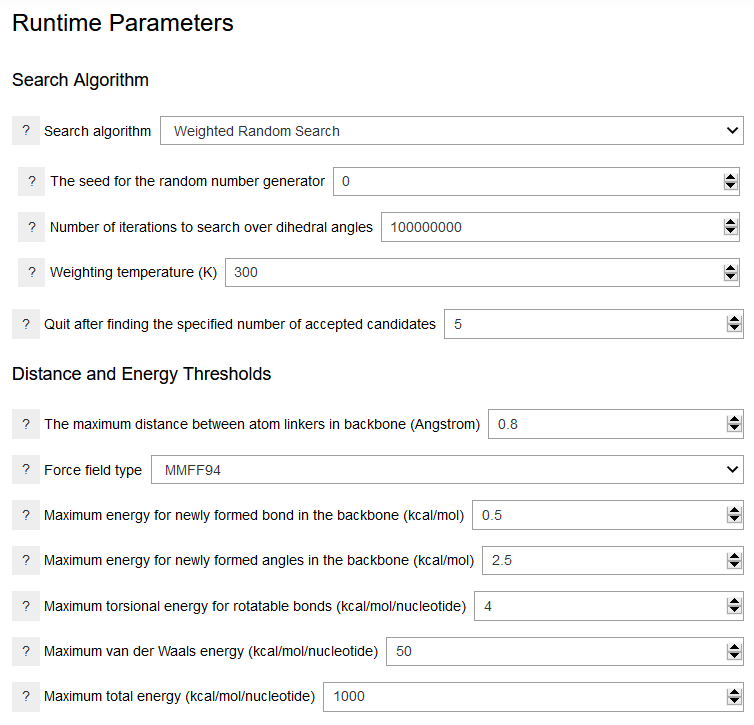
Various conformational search algorithms are available for sapmling the dihedral angles in the backbone. Each algorithm requires specific parameters. For exmaple, the Weighted Random Search algorithm requires values for the number of conformation search steps and the temperature used in the weighting procedure. The random number seed ensures the reproducibility of the run for a given computer platform. Of the available algorithms, only the systematic search algorithm is deterministic and reproducible across platforms. Note that the conformation search procedure is also terminated after the number of candidates specified are accepted. This parameter is set to low values in the examples provided and is encouraged to be adjusted for the production runs. The distance and energy thresholds can be used to refine the generated structures and exclude unreasonable conformers. The distance threshold determines whether the conformer can adopt a periodic structure. It should be set to a small value, such as 0.8 Angstroms. The bond, angle, and torsional energies should also be set to small values. This can accelerate the search and eliminate structures with elongated bonds or strained angles. The van der Waals energy can be used to eliminate structures with significant steric clashes. However, it may vary much depending on the choice of the force field. The total energy also varies significanly with the force field.
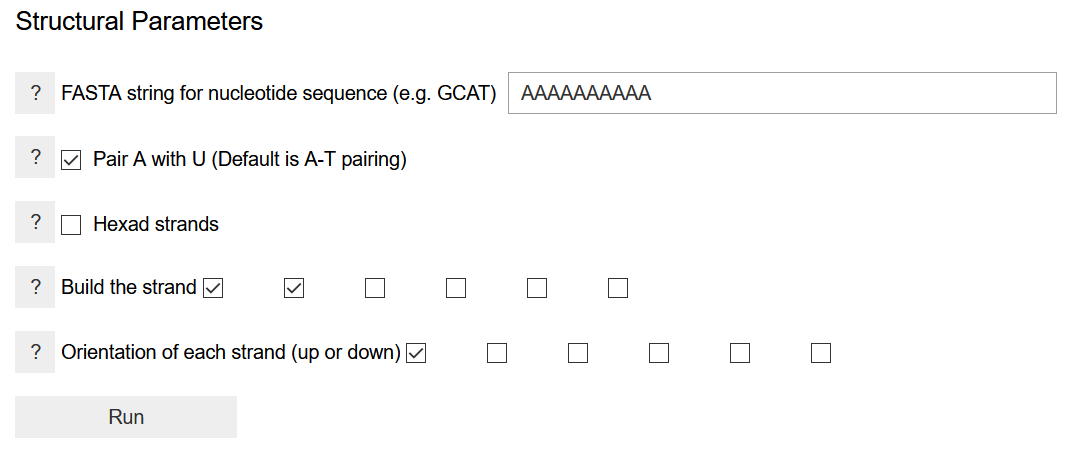
The sequence of nucleobases is written as a FASTA string of the one-letter codes of the nucleobases. It needs to be written for one strand only, while the sequence of other strands is determined by the pairing nucleobase. If building a new system, it might be advisable to start with a short sequence to determine appropriate values for the helical parameters and energy thresholds. Larger strands require more time in energy computation. The default pairing scheme is to pair adenine with thymine. If an adenine-uracil base pairing is wanted, the box should be checked. The program can build hexameric proto-nucleic acid strand. If this is desired, the Hexad strands box should be checked. This tells the program to perform a multiple of 60-degree rotation to generate other strands. By checking on two boxes in the Build the strand option, the program is asked to build a duplex. Clicking on more boxes will build triplexes, quadruplexes, etc. This is appropriate only if the nucleobases are set in the hexad frame of reference, and it does not work for DNA/RNA-like structures. The strand orientation specifies whether to build parallel or anti-parallel strands. The latter is required for DNA/RNA analogs, whereas both options are possible for a hexameric nucleic acid.
The program will use the specified options for running. The time required to complete the run depends on the algorithm options. After it finishes, the program will display all the accepted candidates (if their are any) and their properties.
The results are automatically downloaded as a zip file. Additionally, the output can be downloaded manually by clicking on Download Output. Note that downloading the output is only useful when the program is run in the cloud. When it is run locally the output files are written in the folder where the Jupyter notebook was started.
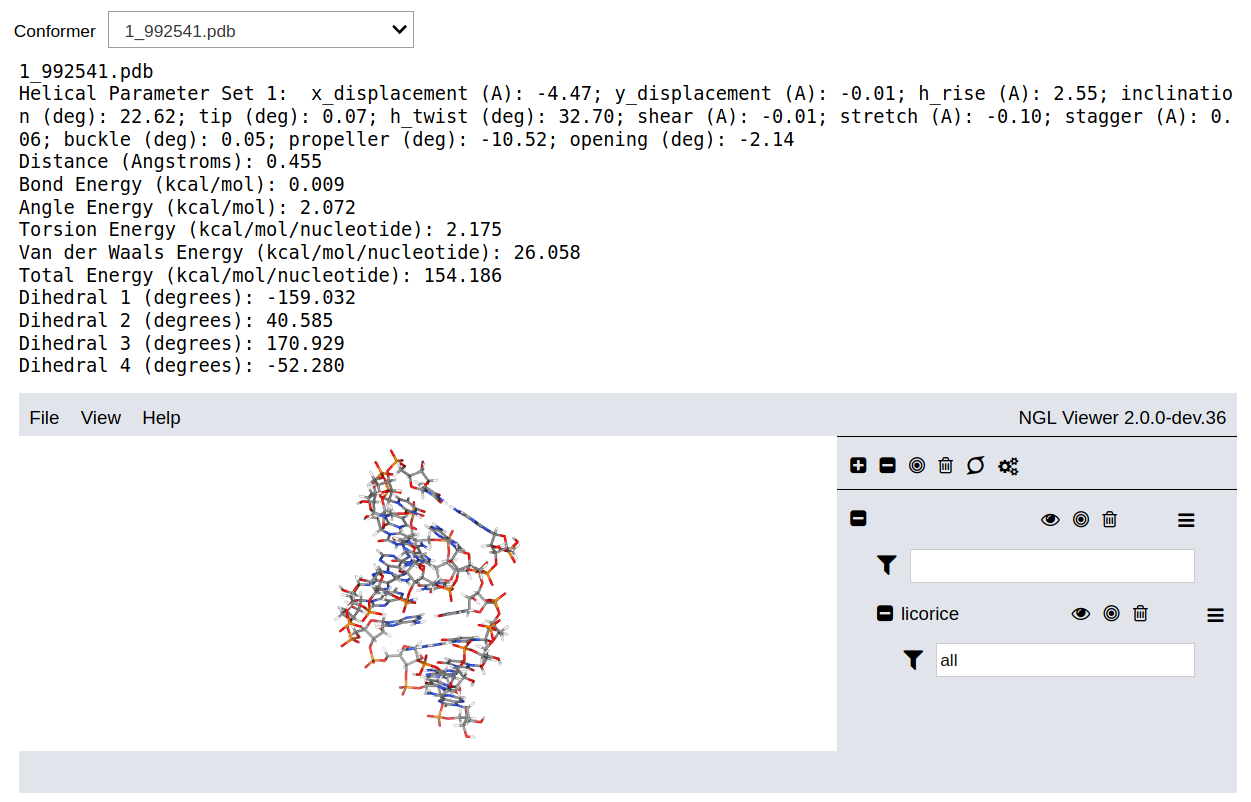
The dropdown list contains all the accepted candidates ordered by the the total energy. The output data are the distance between between one terminal atom of the backbone and the periodic image of the other terminal atom in the adjacent backbone, the energy of the new bond and the new angles formed between two nucleotides, the energy of all rotatable bonds in the backbone, the van der Waals energy, and the total energy. These six terms correspond to distance and energy thresholds defined in the input options. Additionally, the output data include the values of the dihedral angles for the rotatable bonds determined during the conformation search procedure.
Additional Examples
The graphical user interface is essentially a tool for specifying the input options and visualizing the results. However, the functionalities of the program can also be accessed through python scripts.
Basic Structue of the Input File
Here, we show the content of an example options file, which can be written by a text editor to specify the options. Input files can also be generated using the graphical user interface.
Backbone: file_path: RNA_backbone.pdb interconnects: # Backbone to backbone connection - 10 - 1 linker: # Backbone to base connection - 13 - 14
This section specifies the backbone structure. It gives a path to the file containing the three-dimensional structure of the backbone. By default, the program first looks for a file in the current working directory. If it does not find it, it will look for it in the program library. If it does not find it there, it will raise an error. The section also gives the indices of the atoms that connect the backbone to the nucleobase as well as the indices of the atoms that connect backbones together.
An optional entry in this section is specifying fixed rotatable bonds. This can be specified as:
fixed_bonds:
- - 1
- 2
- - 4
- 10
This specifies that the dihedral angle specified by atom indices 1 and 2 and the dihedral angle specified by atom indices 4 and 10 will be fixed during the conformation search.
HelicalParameters: is_helical: true inclination: - 22.9 # Initial point in the range - 22.9 # Final point in the range - 1 # Number of configrations h_rise: - 2.53 - 2.53 - 1 tip: - 0.08 - 0.08 - 1 h_twist: - 32.39 - 32.39 - 1 x_displacement: - -4.54 - -4.54 - 1 y_displacement: - -0.02 - -0.02 - 1
This section specifies the helical parameters. The is_helical keyword determines whether the helical parameters are used or whether the step parameters (i.e. shift, slide, rise, tilt, roll, twist). These are two equivalent schemes for describing the orienation of the nucleobases. When the is_helical value is true, any values specified for the step parameters will not be used. The base pair parameters (shear, stretch, stagger, buckle, propeller, opening) can be similarly specified. The first value is the beginning of the range, the second value is the end of the range, and the third value is the number of configurations in the range. If you want a specific single value, simply specify the same value for both the beginning and the end of the interval.
Note that for hexad geometries, only the rise and the twist are defined. Therefore, the ranges for other entries must be set to zero or removed altogether from the input file.
RuntimeParameters: seed: 0 # Seed for the random number generator search_algorithm: "weighted monte carlo search" # Search algorithm num_steps: 10000000 # Number of steps for backbone dihedral search weighting_temperature: 300.0 # Temperature used in the weighting procedure for dihedral angles monte_carlo_temperature: 300.0 # Temperature used in the Monte Carlo procedure max_distance: 0.8 # Maximum distance between atom linkers in backbone ff_type: GAFF # Force field type energy_filter: - 1.0 # Bond energy involving newly formed backbone bonds - 2.0 # Angle energy involving newly formed backbone bonds - 4.0 # Total torsional energy of rotatable bonds - 0.0 # Total van der Waals energy - 10000000000.0 # Total Energy of the system strand: CGAUUUAGCG # Sequence of base names build_strand: # What strands to build - true # Build one strand only - false - false - false - false - false
This section specifies the runtime parameters for the program. Other search algorithms can be specified as follows:
search_algorithm: "systematic search" dihedral_step: 2
search_algorithm: "monte carlo search" num_steps: 10000000 monte_carlo_temperature: 300.0
search_algorithm: "random search" num_steps: 10000000
search_algorithm: "weighted random search" num_steps: 10000000 weighting_temperature: 300.0
search_algorithm: "genetic algorithm search" num_steps: 1000 population_size: 10000 mutation_rate: 0.75 crossover_rate: 0.25
The example above shows how to build single-stranded RNA by using the keyword build_strand and setting the first value to true and the rest to false. To build a duplex, write:
build_strand: - true - true - false - false - false - false strand_orientation: - true - false - false - false - false - false
For DNA and RNA, the strands are anti-parallel. The strand_orientation option specifies this by setting the first strand to true and the second to false.
To build a hexad,
is_hexad: true build_strand: - true - true - true - true - true - true strand_orientation: - true - true - true - true - true - true
The is_hexad keyword tells the program that we want to use the hexad geometry. The program, therefore, will perform 60 degrees rotation when appropriate to generate the strands. To build all the six strands in the hexad, set all entries in build_strand to true. The strand_orientation option allows you to build any cobmination of parallel and anti-parallel strands by setting the values to true or false
Adding Bases Not Defined in The Library
The program can build structures with arbitrary nucleobases. The key problem with defining a new nucleobase is making sure that the provided geometry of the nucleobase is in the correct standard frame of reference. Otherwise, the helical parameters will be ill-defined. The additional bases can be defined in the graphical user interface or by editing the input file. The program can automatically align provided nucleobases to the standard DNA/RNA frame of reference, though the user may need to verify that the nucleobase is correctly aligned. The program cannot align bases to the hexad frame of reference.
For the generated strands, the length of the bond between the nucleobases and the backbone is determined by the bond length of the first nucleotide in the strand. The bond length is determined by the van der Waals radii of the bonded atoms. It can also be set manually by the user.
The program has a library of predefined nueleobases. They include adenine, guanine, cytosine, thymine, and uracil. Additionally, there are two proto-nucleobases that are used for building the hexads, namely aminopyrimidine and cyanuric acid. To define other nucleobases, add the following entries to the input file:
Base modified_adenine: code: AD2 # Three letter PDB code file_path: modified_adenine.pdb # Path to the 3D structure of the nucleobase linker: # Indices of the vector forming the bond between the nucleobase and the backbone - 5 - 11 name: Q # One-letter name of the base pair_name: T # One-letter name of the pairing base
These lines will add the nucleobase modified_adenine to the library of available nucleobases. For the name of the base, you must not use any of the names already used in the library. Otherwise, your new base might be overwritten. The reserved names are the following: A, G, C, T, U, I, and E. These names are case-insensitive.
Now that you defined a new nucleobase, you can use its name when you specify the strand:
strand: CGCQUQTGQG
You can define additional nucleobases in the same way.
Adding Files to the Library
The library of the program is in the folder data in the package folder. The library contains example files and the coordinates for the defined bases and backbones. You can add additional files there to make them accessible wherever you run your Jupyter notebook or python script. To add other nucleobases to the library, edit the bases_library.yaml files.
Python Script
The minimal input to run the program as a python script is this:
By default, the program first looks for the file RNA.yaml in the current working directory. If it does not find it, it will look for it in the program library. If it does not find it there, it will raise an error. This creates an instance of the driver.pNAB class, given the options specified in the input file, and runs the program.
To access the options programmatically, you can use the options attribute:
This attribute is a dictionary of all the defined options, and it can be changed in the script.
After running the program, the instance stores all the results in the results attribute.
This is a numpy array containing the information about the accepted nucleic acid candidates, that is the candidates that satisfied the distance and energy criteria. The columns in the array are defined accroding to the corresponding entry in the header attribute. The first column in the results contain the prefix value. This is an integer that indicates the sequence of the helical configuration. This parameter is useful when you run a range of values for the helical parameters, where each helical configuration would have a different prefix. The prefix attribute is a dictionary whose keys are strings of the sequence of integers and whose values are strings specifying the corresponding helical configurations.
When multiple helical configurations are defined, e.g. by giving a range of values for the twist angle and asking for 5 configurations, the program runs these different helical configurations in parallel. The number of calculations that can be run in parallel depends on the available processors in the computer. By default, the program uses all available CPUs for parallel calculations. To use a different number, you can pass a keyword argument to the run function:
Additionally, the program prints a progress report to the screen by default. If you do not want the progress report, type:
